Understanding Cancer’s Growth and the Advancements of Oncological Treatments for Various Cancer Types

According to a scientific report published in 2021, the worldwide cancer burden in developing nations is expected to climb by 47% between 2020 and 2040. The number is worrying since new globalisation-related issues are exacerbating demographic changes (Sung et al., 2021). As a result, continued oncological research is required to have a more comprehensive understanding of the molecular pathways that lead to cancer.
Cancer is a complex, heterogenous defect of genetic origin. The first objective of this essay aims to provide a thorough synopsis on the advancement of cancer from DNA damage, early benign lesions to genetically heterogeneous malignant tumours, utilising specific cancer types as examples, whilst exploring current and future antineoplastic treatments. Cellular mechanisms that drive cancer initiation, promotion, progression, malignancy, and metastasis will be critically analysed. The second objective of this essay will explore the current and future antineoplastic treatments used to treat cancer, focusing on how this knowledge can inform the design of future treatments, whilst exploring genetic heterogeneity; the characteristic of cancer cells within the same tumour can possess drastically distinct mutations. Undoubtedly, this would affect how one’s treatment plan is structured, even if two patients were afflicted with identical cancer types. By achieving these objectives, this essay aims to contribute to a more comprehensive understanding of cancer development and the advancement in efficacious antineoplastic treatments. The significance of this topic is underscored by the alarming statistics of rising cancer rates, with female breast cancer surpassing lung cancer as the most commonly diagnosed cancer globally, estimated at 2.3 million new cases in 2020 (Sung et al., 2021). Therefore, it is crucial to gain further insight into the stages which enable its transition.
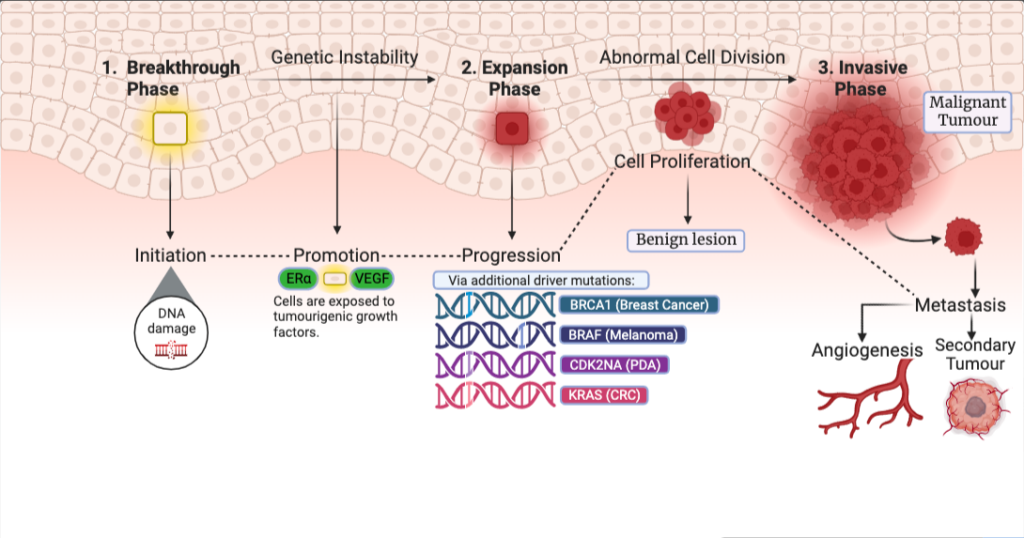
Figure 1: Phases of cancer: which move cancer from a benign lesion to a complete malignant tumour; breakthrough phase consisting of driver mutations which initiates the DNA damage to the expansion phase consisting of additional driver mutations which leads to the formation of a benign and the final transition into a malignant tumour. The accumulation of mutations, subsequent driver mutations and formations of benign lesions to malignancy can take decades to occur. Figure 1 was created in BioRender.
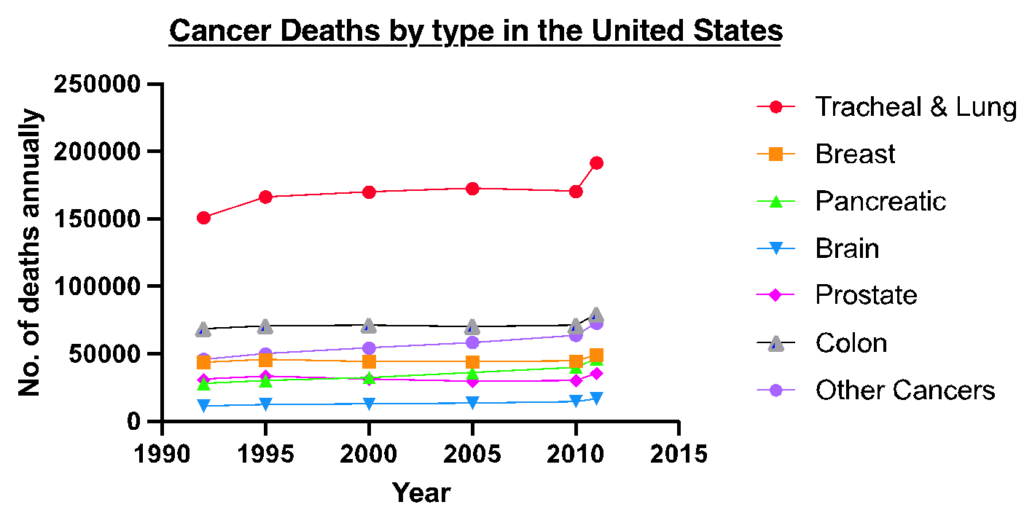
Figure 2: Cancer fatalities in the United States: The graph illustrates the specific cancer types which have been increasing in their mortality rates over the last 25 years. Some cancers did show an initial increase (i.e., breast cancer) but then increased in the number of fatalities towards 2015. For others such as lung cancer, fatalities have been increasing due lifestyle factors such as smoking and being exposed to 2nd hand smoke. Other cancers are comprised of cancer with a death toll lower than <100,000 including nasopharynx, testicular and uterine cancers. Figure was created in GraphPad Prism. Data obtained from Max Roser and Hannah Ritchie, 2019.
1. Initiation (Breakthrough Phase) – DNA damage resulting in genomic instability:
The initiation of malignancy is always of genetic origin. It arises via intrinsic DNA damage (i.e., malfunctions in DNA repair and replication mechanisms or expulsion of free radicals, a by-product of cellular metabolism, resulting in oxidative DNA damage) (Alhmoud et al., 2020). Conversely, extrinsic DNA damage (i.e., mutagenesis caused by carcinogens such as tar or exposure to high-frequency wavelengths like gamma rays which cause direct DNA damage) also causes DNA damage which will be the primary focus of this section. This DNA damage will result in genomic instability, resulting in genetic aberrations; driver mutations that will bring about the breakthrough phase of a cancer. A notable example is lung cancer. The inhalation of cigarette smoke will expose respiratory epithelium to carcinogens i.e., polycyclic aromatic hydrocarbons (PAHs) such as Benzo[a]pyrene. These PAHs metabolise into electrophiles which bind to nitrogenous bases covalently, resulting in the destabilisation of N-Glycosyl bonds causing depurination of DNA bases. This leads to the generation of DNA adducts. The majority of DNA adducts will be repaired by repair mechanisms (G1/M-Phase checkpoint), though some may evade repair. These DNA adducts will induce driver mutations in genes (Ma and Li, 2017). This pathway is visualised in figure 3. Entire DNA bases can be lost as a result. If these alterations become ‘locked in’ within the cell cycle via DNA repair mechanisms it can result in detrimental genetic aberrations (Casale et al., 2001). Moreover, the accumulation of these DNA adducts can induce mutations within oncogenes, initiating tumour carcinogenesis (Ceppi et al., 2017). Another form of extrinsic DNA damage is deamination; carcinogens chemically modify bases resulting in the complete change of a base E.g., the pyrimidine cytosine is converted into uracil. Once this mutation becomes ‘locked in’ via DNA repair mechanisms, this genetic error will become a permanent addition to an individual’s genetic code. If a cell with this genetic anomaly enters the cell cycle without being countered by apoptotic mechanisms, it results in the accumulation of further driver mutations. This highlights the breakthrough phase potentially leading to the formation of a benign lesion within lung epithelia. Mutations like this can vary from cell to cell, leading to tumour heterogeneity. Lung cancers exhibit distinct genetic profiles, resulting in varying reactions to the same treatment. Understanding the genetic variability of lung cancer can aid in the development of innovative antineoplastic treatments.
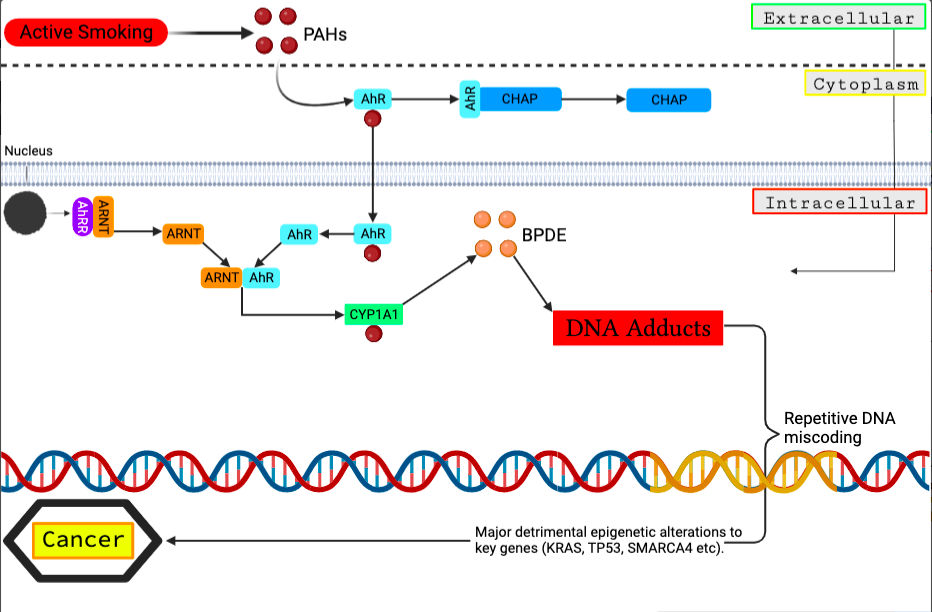
Figure 3 – Molecular pathway of external mutagenesis causing DNA adduct formation: This cascade is initiated by the inhalation of cigarette smoke containing PAHs which penetrate pulmonary cells and attach to the AhR transcription factor. As a result, CYP1A1 is expressed, and PAHs are metabolised into hydrophilic intermediates that can be detoxified or generate DNA adducts. Changes in methylation of CYP1A1 or AhRR enhances CYP1A1 expression and generate more DNA adduct formation, resulting in DNA miscoding. Long-term smoking can result in persistent DNA miscoding and epigenetic alterations in cancer genes. Figure created in BioRender, adapted from Ma and Li, 2017.
1.1 ICB Immunotherapy for lung adenosarcomas:
Damaged lung cells take advantage of cell cycle checkpoints, evading detection whilst remaining locked within the cycle, acquiring more detrimental mutations, accumulating more DNA damage (E.g., G1 checkpoint will search to destroy cells containing DNA adducts but damaged cells are able to evade such checkpoints by accumulation of mutations to key Tumour Suppressor Genes which regulate cell cycle checkpoints). Immune checkpoint blockade (ICB) therapy involves utilising pharmaceutical intervention to block the cell cycle checkpoints enabling the immune system to recognise and destroy. Predicting the responses to ICB is challenging, as 70% of patients cannot withstand a high mutational burden. These early driver mutations lead to processes of depurination and deamination, forming DNA adducts. Tertiary lymphoid structures (TLS) adjacent to tumours are regions abundant in B & T cells, a strong indicator of an ICB response within lung adenosarcoma. Another indicator of damaged cells are cancer-associated antigens; Endogenous Retrovirus Glycoproteins (ERV). The purpose of this study was to combine the effects of TLS and ICB to target lung adenocarcinomas. Specifically utilising B cells that perceive ERV as dominant antibody targets. A mouse model demonstrated that local and systemic anti-tumour B cell responses could develop anti-tumour immunity via the secretion of antibodies which specifically target tumour-associated antigens, such as ERV, Experimental analysis is expanded upon in figure 4. This is coordinated by introducing immunotherapy to combine the TLS and ICB to work in conjunction. Furthermore, the outcomes of this murine model strongly support this treatment as boosting anti-tumour antibody responses by ICB was indicative of a broader of PD-1/PD-L1, keeping the damaged cells from evading cell cycle checkpoints. What is significant about this treatment is that both TLS and ICB function cooperatively to block damaged cells at cell cycle checkpoints preventing entry into the cycle. B cells destroy these damaged cells. These effects have been observed in the humoral response to both foreign and self-antigens (Ng et al., 2023). This highlights an antineoplastic treatment to treat lung adenosarcomas in its earlier stages.
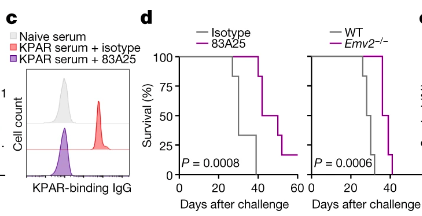
Figure 4 – Survival of in vivo murine models when exposed to 83A25: Graph C visualises 83A25 treatment presenting a similar trend to naïve serum treatment Graph D depicts the survival of KPAR tumour-bearing mice treated with 83A25 or isotype control antibodies, as well as untreated wild-type and Emv2/ hosts. The findings suggest that 83A25 therapy can increase survival in KPAR tumour-bearing mice. Figure adapted from Ng et al., 2023.
2. Promotion – Growth factors acting on initiated cells causing selective proliferation:
The promotion stage of tumorigenesis consists of the initiated cells which have undergone DNA damage resulting in genetic abnormalities. The unique aspect of this stage is the ability-initiated cells obtain; the capacity to divide into preneoplastic lesions – benign neoplasms. The initial mutation occurring within the breakthrough phase, makes the cells susceptible to more mutations; a unique attribute of the promotion stage leading to the formation of benign lesions (Compton, 2020). They have accumulated as the cell has evaded the repair checkpoints throughout the cell cycle. These acquired mutations, genetic alterations and exposure to specific growth factors provide a selective growth advantage within the initiated compared to the neighbouring cells. ER+ breast cancers demonstrate this molecular mechanism. Oestrogen Alpha Receptor (ERα) is a transcription factor which is activated by oestrogen in breast tissue. This transcription factor possesses the ability to awaken dormant oncogenic pathways triggering genes such as Cyclin D1 or HER2; oncogenes notorious for their role in unregulated cell growth and proliferation. Cyclin D1 protein is a crucial regulator of the cell cycle. If these unregulated mitotic divisions are allowed to persist, then the outcome leads to the formation of a benign lesion within the breast tissue.
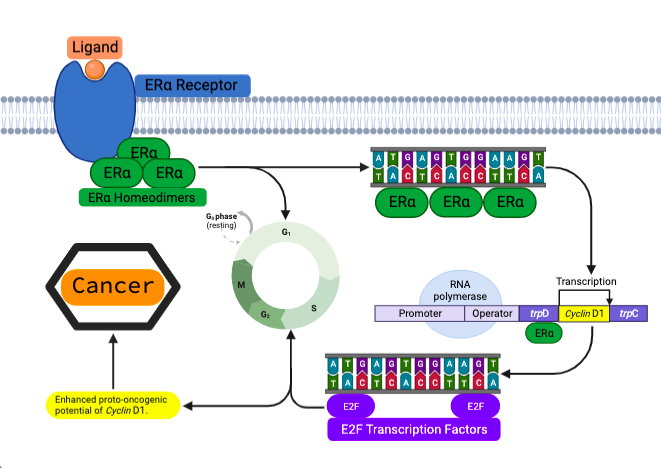
Figure 5 – Molecular pathway of Cyclin D1 activation via ERα: Oestrogen will bind to ERα receptors activating its transcriptional capability. ERα protein takes form in homodimers binding to specific DNA sequences. This will be to regulate the expression of certain genes, one gene being Cyclin D1. Cyclin D1 will form a complex with E2F. This liberates E2F transcription factors causing the cycle to progress into the G1/S Phases. This promotes cell proliferation and growth, enhancing the oncogenic potential of Cyclin D1. Figure created in BioRender, adapted from Bartek and Lukas, 2011.
2.1 YAP inhibition of ERα to disrupt ERα pathways disrupting breast sarcoma formation:
A novel treatment presented by Li and colleagues involves Yes, Associated Protein (YAP), a transcriptional co-activator which controls cell proliferation. A specific pathway controls cell growth, proliferation, and apoptosis within cells; HIPPO pathway. Within its default setting, YAP activity is suppressed. Insight into this this pathway uncovered that if the cascade was disrupted (i.e., by a potent Large Tumour Suppressor Kinase inhibitor), YAP accumulates within the cytoplasm. YAP binds to TEAD; a family of highly conserved transcription factors, via pharmaceutical intervention (specific kinase inhibitors). TEAD is a mandatory cofactor for ERα, it physically associates to ERα activating its oncogenic traits; promoting ERα, binding to its target enhancers. YAP will bind to TEAD, altering its structure. TEAD can no longer bind to, thus utilise the super enhancer to then interact with ERα, suppressing the oncogenic capability of ERα. This disrupts the ER dependent breast sarcoma growth (Li et al., 2022). Said treatment provides an outlook toward a potentially efficacious antineoplastic treatment which halts the promotion phase of cancer from leading to progression, halting the formation of benign lesions.
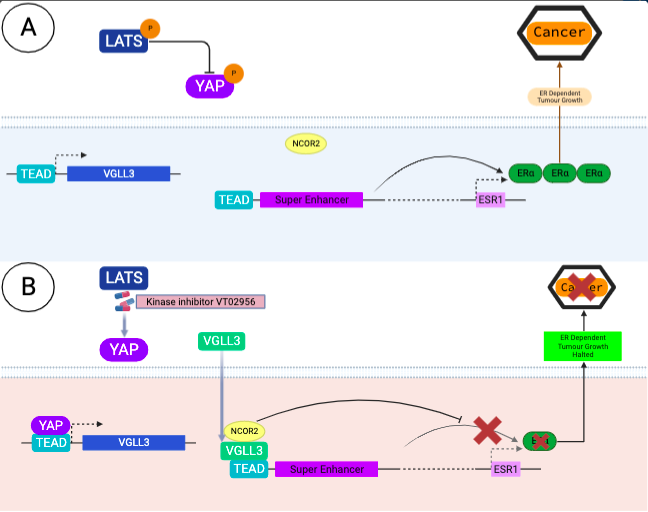
Figure 6 – Proposed model of molecular action of YAP on TEAD with VGLL3 and kinase inhibition: A: Details the ordinary progression of ER Dependent tumour growth with the expression of ERα. B: Details the pharmacological inhibition of LATS with a kinase inhibitor VTO2956, which blocks TEAD from interacting with its super enhancer in its ordinary capacity. This prevents the activation of ERα which cannot mediate its oncogenic ER dependent pathways. This shows the importance of antineoplastic treatments in the promotion phase of cancer; Promotion is reversible and can be halted with the correct treatment. Figure created in BioRender, adapted from Ma et al., 2022.
3. Progression (Expansion Phase) – accumulation of additional mutations driving preneoplastic, benign lesions to acquire a malignant phenotype:
Progression of tumorigenesis is differentiated from the promotion stage. An abundance of the driver mutations mentioned prior in key genes (Tumour Suppressor Genes/TSGs) have been acquired to where the genetic abnormalities are non-reversible. For instance, the p53 TSG is mandatory in regulating the cell cycle. A mutation within p53 could partially disrupt its function in regulating cell cycle checkpoints in G1 & S-Phase (cell proliferation checkpoints) keeping the damaged, damaged cells locked within the cell cycle, specifically, instead of forcing them to enter senescence. Here, the damaged cells will evade apoptosis mechanisms. (Ou and Schumacher, 2018). Further driver mutations will disrupt the function of p53 completely, rendering the gene functionless. In melanoma, these mutations will over-express AXL; a receptor tyrosine protein kinase involved in MDMX-MDM2 pathway, which regulates the transcriptional activity and protein production of p53. Both protein homologues contain RING domains which activates the enhancement E3 ligase causing MDM2 to bind to the transactivation domain of p53, suppressing its transcriptional activity. Regardless of cutaneous melanoma having one of the lowest incidence rates of p53 mutation the expression of AXL will stabilise the MDMX-MDM2 pathway, suppressing p53 function, a crucial step in the cancer progression; the outcomes being heightened levels of cell proliferation, continuation of genomic instability and G1 checkpoint failure (de Polo et al., 2017). This enables replicative immortality, progressing the cancer onwards from the benign lesion, expansion phase. Hanahan and Weinberg denoted replicative immortality as one of the hallmarks of cancer (Hanahan and Weinberg, 2000). Accumulation of driver mutations from initiated ones alongside consistent DNA damage can take decades to culminate into advanced stages of cancer. An advanced stage of cancer development could culminate in an invasive malignancy.
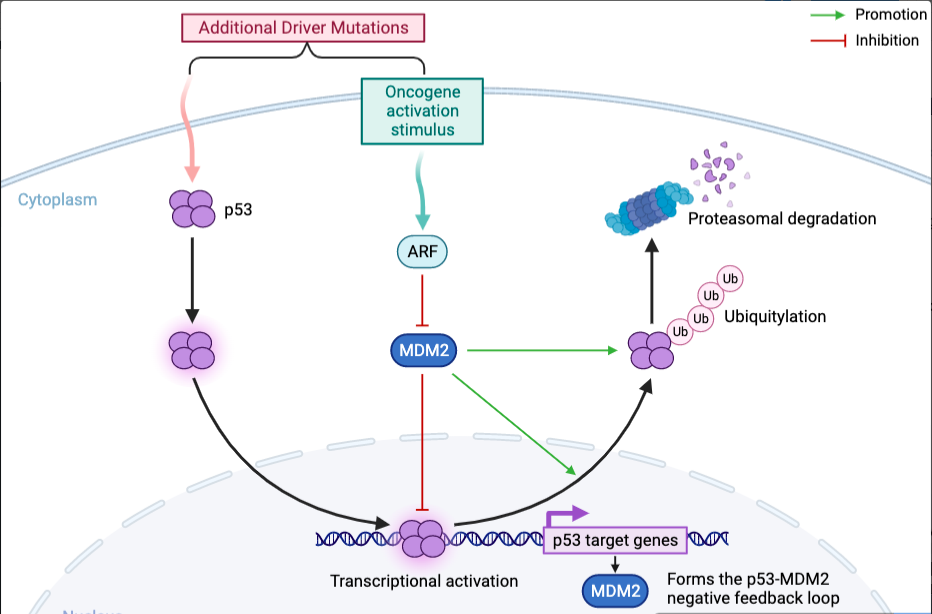
Figure 7 – Molecular action of p53 suppression via the MDMX-MDM2 pathway: MDM2 is an oncogene that can cause cancer when it’s mutated. The figure illustrates the interaction between three proteins – p53, MDM2, and ARF – in the context of cancer. The MDM2-p53 complex regulates p53’s tumor-suppressive functions. Disrupting the MDM2-p53 interaction activates p53 and induces MDM2 expression through negative feedback. ARF promotes p53 activation by inhibiting MDM2. Degradation of the proteasome completely disrupts p53’s ability to suppress tumors. This figure was adapted from Kung and Weber (2022) using BioRender.
3.1 Inhibition of AXL increases expression of p53 target genes in melanoma cells:
Pharmaceutical AXL inhibitors were examined in A375 melanoma cells to determine their method of action, effectiveness, and p53 consolidation. Their findings revealed promising results in the stabilisation of p53, whilst also indicating a rise in protein synthesis of p53 target genes p21 and PUMA. This provides a potentially effective therapy option for restoring p53 to its native role as a crucial gene in tumour suppressor activity (de Polo et al., 2017).
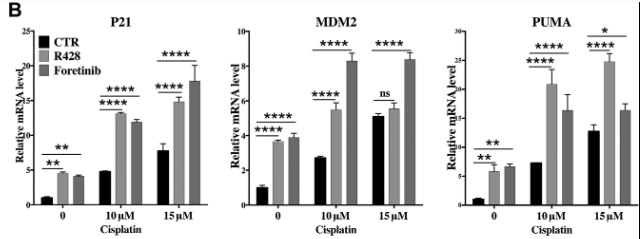
Figure 8 – Enhancement of p53 pathway via Cisplatin treatment: Results showed that A375 cells were taken and had AXL silenced by siRNA. Cells were treated with cisplatin to assess the response of p53 target genes. Upon treatment MDM2 protein expression increased whilst MDMX levels decreased via MDM2 mediated degradation of MDMX. Graphs demonstrate PUMA and p21 protein increase, target genes p53 protein. Effects were further enhanced by AXL silencing. Adapted from (de Polo et al., 2017).
4. Malignancy (Invasive Phase) – Cells have gained the ability to invade adjacent tissues whilst possessing rapid, unregulated growth.
Malignancy differs from the progressive phase in that it has aberrant nuclei (in the form of aneuploidy), uncontrolled cell proliferation, and the potential to penetrate neighbouring tissues, which benign lesions do not have. Because of anomalies in the cell cycle, these aberrant cells sustain this high pace of uncontrolled mitosis. One study focuses on how the protein kinase CDK1 triggers cell cycle entrance. Cyclin B1 protein, which forms a complex with CDK1 to promote G2 to M-Phase development, regulates CDK1. In cancer cells, due to the accumulation of genetic aberrations, Cyclin B1 becomes activated as an oncogene, with Cyclin B1 protein being overproduced. CDK1 is overstimulated via direct binding of Cyclin B1, causing the cycle to progress onwards whilst the Cyclin B1 – CDK1 complex is active, bypassing G2 to M-Phase checkpoints via phosphorylation; Wee1 and CDC25 phosphatase. Wee1 and CDC25 phosphatase cannot initiate cell cycle arrest nor drive the cells into senescence. Hence, failure of apoptotic regulatory mechanisms sustains replicative immortality, causing invasion within neighbouring tissues (Helen K. Matthews et al., 2021); one of the hallmarks of cancer presented by Weinberg and Hanahan (Hanahan and Weinberg, 2000).
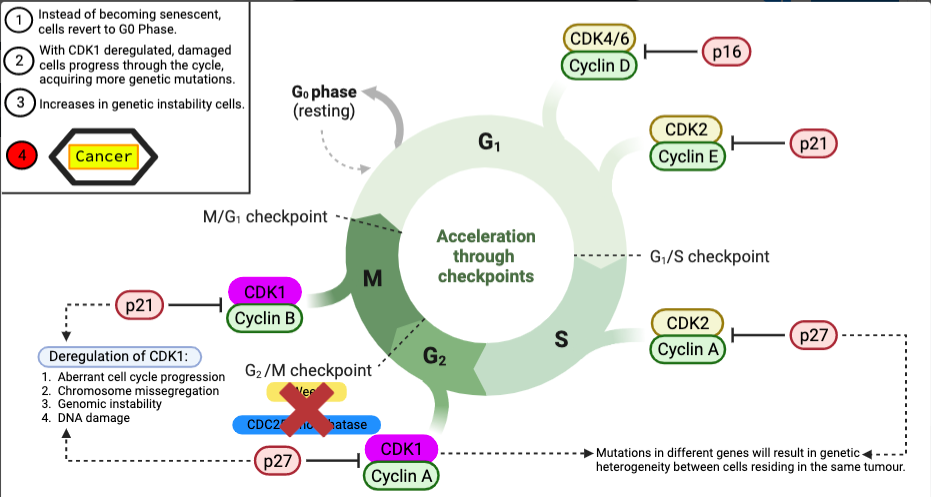
Figure 9 – Deregulation of CDK1: Wee1 and CDC25 phosphatase regulate CDK1, which is required for normal cell cycle progression. CDK1 is phosphorylated and inhibited by Wee1, whereas CDK1 is dephosphorylated and activated by CDC25. Deregulation of CDK1 activity increased Wee1 activity leads to extended cell cycle progression and genomic instability, resulting in malignancy. CDK1 dysregulation can result in genetic heterogeneity in cancer by allowing chromosomal abnormalities to accumulate. As cells divide uncontrollably as a result of genomic changes, they can give rise to subclones of cells with different mutations, resulting in a variety of cancer cells within a tumour. Different cells respond differently to therapies, tumours difficult to treat. Figure created in BioRender.
4.1 Utilising BMAL1 as a therapeutic target to treat glioblastomas:
A highly lethal example of malignancy would be glioblastomas. They are categorised as the most malignant tumour of the nervous system. A study has uncovered since 2005, 5-year survival rates have not improved (Poon et al., 2020). A novel therapeutic treatment looks to target BMAL1; a transcription factor which is primarily associated with regulation of circadian rhythms. BMAL1 is also implicated in the regulation of the cell cycle, specifically the G1 to S-Phase transition. The study describes the increase of Cyclin B1 causing a decrease in the presence of BMAL1 in tumour cells. A decrease in BMAL1 within cell results in increased cell proliferation, growth, along with invasion of adjacent tissues, whereas an increase in BMAL1 will present the opposite effect: inhibition of cell proliferation and movement of malignant cells to invade neighbouring tissues. The in vivo protocol utilising U87MG cells conducted by Gwon and associates disclosed that Cyclin B1 exposure decreased BMAL1 presence. Results showed an increase of 24% increase in cell number of post G1 cells, compared to when the same cells were exposed to the negative control. Moreover, adenovirus mediated ectopic expression of BMAL1 within U87MG cells demonstrated a decline in cell migration (Gwon et al., 2020). These findings are in support of a promising antineoplastic treatment which could treat glioblastomas.
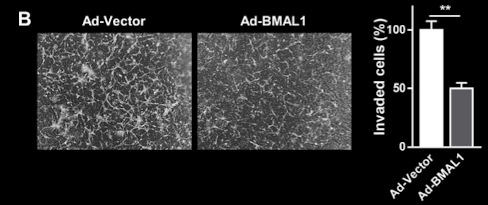
Figure 10 – Overexpression of BMAL1 reduces cell migration and invasion: Figure B demonstrate that overexpression of BMAL1 in U87MG cells resulted in a significant decrease in the number of invaded cells compared to control cells. A micrograph displays a significant decrease in cell migration in the Ad-BMAL1 compared to the Ad-Vector. Figure adapted from Gwon et al., 2020.
5. Metastasis: Movement of malignant cells to distant tissues to initiate a secondary neoplasm.
Metastasis refers to the detachment of tumour cells from their primary site, movement throughout the bloodstream or lymphatic system to establish a secondary tumour at a distant tissue or organ. One prominent cancer known for metastasis would be pancreatic ductal adenocarcinoma (PDAC); Metastatic disease is already found in most patients at time of diagnosis. Ultimately, leading to a 5-year survival rate below 5% (Bengtsson, Andersson and Ansari, 2020). Usual sites of secondary tumour propagation include the peritoneum at 48% and lungs at 45%. Patients will present these sites of metastases 76-80% upon diagnosis (Miquel, Zhang and Pilarsky, 2021). Essential processes need must for metastasis to occur; degradation of the basal lamina, movement of tumour cells from the primary tumour site via the bloodstream or lymphatic system to the secondary site & establishment of the tumour microenvironment etc. This is expanded upon in figure 11.
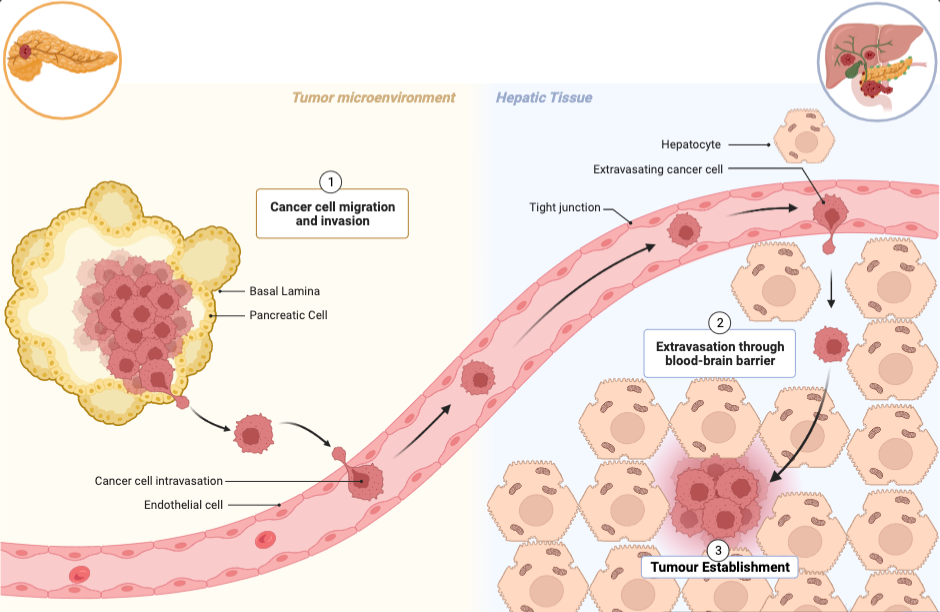
Figure 11 – The stages of metastasis which lead to secondary tumour site from PDAC to liver: Tumour cells will accumulate at the basal lamina of pancreatic tissue. At this stage, the cancerous neoplasm is removable via surgery. However, if the tumour cells are able to degrade the basal lamina by releasing proteases, the tumour cells will be released into the bloodstream or lymphatic system. These tumour cells will induce extravasation out of the bloodstream into the hepatic tissue to establish a tumour microenvironment. Eventually the tumour will move from cold to hot, restructuring the immune cells surrounding the tumour which amplify inflammation. From this point onwards, the tumour can commit to angiogenesis. Figure adapted in BioRender, adapted from Julien Cicero (Creator) & Erin Marshall.
5.1 Pursuing TAMS as therapeutic targets to cease pancreatic angiogenesis:
PDAC possesses one of the highest rates of cancer metastasis of all cancer types; One study reported that 70% of patients had developed distant tissue metastasis, 84.3% showing liver metastasis specifically (Tao et al., 2016). PDAC is also the third cause of cancer related death in the US and is estimated to be the second by 2030 (Dimitrov-Markov et al., 2020). A recent therapeutic study identified the tumour microenvironment coordinated a significant role in secondary tumour site establishment of PDAC; specifically, tumour associated macrophages (TAMs) aiding with the development of angiogenesis. Hanahan and Weinberg also identified angiogenesis as a hallmark of cancer (Hanahan and Weinberg, 2000). Analysis of the in vitro protocol showed M2 macrophages synthesised MDEs; miniscule vesicles were able to generate new blood vessels within Pan02 cell-derived exosomes. These MDEs contained proteins which promote blood vessel formation within PDCA. Yang and colleagues discovered that endothelial cells uptake MDEs, enhancing their ability to execute angiogenesis. The results of the in vivo protocol confirmed their previous findings.
Researchers at PDCA tumour centres injected M2 macrophages containing MDEs into their murine models. The findings strongly suggested that M2 MDE macrophages increased cell proliferation and angiogenesis in mice, as evidenced by a larger tumour mass and greater micro vascular density (MVD) compared to the negative control. TAMs, both in vitro and in vivo, stimulate angiogenesis via MDEs. To treat metastasis, a therapeutic target for both the macrophage and the vesicle can be identified (Yang et al., 2021). However, because oncologists do not fully understand the mechanisms underlying the tumour microenvironment (Quaranta et al., 2018), more research into future antineoplastic treatments is required. Specifically, when tumours have progressed to this perilous stage, metastasis.
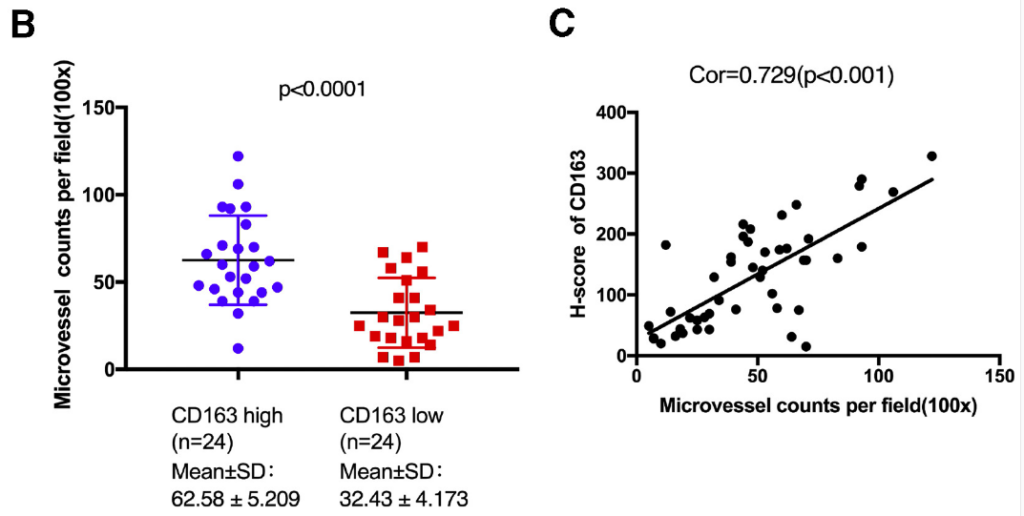
Figure 12: MVD increase upon exposure to M2 Macrophages containing MDEs within In vivo models containing tumours: Graph B details the increase of MVD compared to the negative control, due to tumours within the murine models being exposed to M2 macrophages and MDEs, promoting novel blood vessel formation. Graph C reinforces finding as the H Score detected an increase of MVD formation as MDEs were introduced. This details how M2 macrophage presence promotes angiogenesis, identifying a target for therapeutic treatment of metastasis. Figure obtained Yang et al., 2021.
6. Conclusion
In summary, this essay has detailed how the accumulation of DNA damage within cells will form a benign lesion. If these genetic aberrations continue to persist without apoptotic mechanisms dismantling preneoplastic cells, then a malignancy will arise. Each of the cancers utilised in this literature demonstrate why a universal cancer treatment is unfeasible; Genetic heterogeneity. Currently, over 200 cancers human cancers have been identified (Cancer Research, 2020), excluding cancer subtypes. Even if two patients were diagnosed with identical carcinomas, the genetic disparities between the patients requires personalised treatments. Other factors such as age, lifestyle, gender, response to current treatment etc will also impact how a patient’s treatment is structured. Moreover, if a specific therapy indicates efficacious results, there is no guarantee the same treatment will provide similar results in the promotion phase even in the same cancer subtype. Hence, why continued research into understanding tumour heterogeneity will be compulsory in developing future antineoplastic treatments for various cancer types.
7. Bibliography
Alhmoud, J.F., Woolley, J.F., Al Moustafa, A. and Malki, M.I. (2020) ‘DNA damage/repair management in cancers’, Cancers, 12(4), pp. 1050.
Bartek, J. and Lukas, J. (2011) ‘DNA repair: Cyclin D1 multitasks’, Nature (London), 474(7350), pp. 171.
Bengtsson, A., Andersson, R. and Ansari, D. (2020) ‘The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data’, Scientific Reports, 10(1), pp. 16425.
Cancer Research, U.K. (2020) Types of cancer. Available at: https://www.cancerresearchuk.org/what-is-cancer/how-cancer-starts/types-of-cancer#:~:text=For%20example%2C%20nerves%20and%20muscles,breast%20cancer%20or%20lung%20cancer. (Accessed: .
Casale, G.P., Singhal, M., Bhattacharya, S., RamaNathan, R., Roberts, K.P., Barbacci, D.C., Zhao, J., Jankowiak, R., Gross, M.L., Cavalieri, E.L., Small, G.J., Rennard, S.I., Mumford, J.L. and Shen, M. (2001) ‘Detection and quantification of depurinated benzo[a]pyrene-adducted DNA bases in the urine of cigarette smokers and women exposed to household coal smoke’, Chemical Research in Toxicology, 14(2), pp. 192-201.
Ceppi, M., Munnia, A., Cellai, F., Bruzzone, M. and Peluso, M.E.M. (2017) ‘Linking the generation of DNA adducts to lung cancer’, Toxicology (Amsterdam), 390, pp. 160-166.
Compton, C. (2020) ‘Cancer initiation, promotion, and progression and the acquisition of key behavioral traits’Cancer: The Enemy from Within Switzerland: Springer International Publishing AG, pp. 25-48.
de Polo, A., Luo, Z., Gerarduzzi, C., Chen, X., Little, J.B. and Yuan, Z. (2017) ‘AXL receptor signalling suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex’, Journal of Molecular Cell Biology, 9(2), pp. 154-165.
Dimitrov-Markov, S., Perales-Patón, J., Bockorny, B., Dopazo, A., Muñoz, M., Baños, N., Bonilla, V., Menendez, C., Duran, Y., Huang, L., Perea, S., Muthuswamy, S.K., Al-Shahrour, F., Lopez-Casas, P.P. and Hidalgo, M. (2020) ‘Discovery of new targets to control metastasis in pancreatic cancer by single-cell transcriptomics analysis of circulating tumor cells’, Molecular Cancer Therapeutics, 19(8), pp. 1751-1760.
Hanahan, D. and Weinberg, R.A. (2000) The hallmarks of cancer Elsevier Inc.
Helen K. Matthews, Cosetta Bertoli and Robertus A. M. de Bruin (2021) ‘Cell cycle control in cancer ‘, (January 2022).
Kung, C. and Weber, J.D. (2022) ‘It’s getting Complicated—A fresh look at p53-MDM2-ARF triangle in tumorigenesis and cancer therapy’, Frontiers in Cell and Developmental Biology, 10, pp. 818744.
Li, X., Zhuo, S., Zhuang, T., Cho, Y.S., Wu, G., Liu, Y., Mu, K., Zhang, K., Su, P., Yang, Y., Zhang, C.C., Zhu, J. and Jiang, J. (2022) ‘YAP inhibits ERα and ER+ breast cancer growth by disrupting a TEAD-ERα signaling axis’, Nature Communications, 13(1), pp. 3075.
Ma, S., Tang, T., Probst, G., Konradi, A., Jin, C., Li, F., Gutkind, J.S., Fu, X. and Guan, K. (2022) ‘Transcriptional repression of estrogen receptor alpha by YAP reveals the hippo pathway as therapeutic target for ER+ breast cancer’, Nature Communications, 13(1), pp. 1061.
Ma, Y. and Li, M.D. (2017) ‘Establishment of a strong link between smoking and cancer pathogenesis through DNA methylation analysis’, Scientific Reports, 7(1), pp. 1811-13.
Max Roser and Hannah Ritchie (2019) ‘Cancer’, .
Miquel, M., Zhang, S. and Pilarsky, C. (2021) ‘Pre-clinical models of metastasis in pancreatic cancer’, Frontiers in Cell and Developmental Biology, 9, pp. 748631.
Ng, K.W., Boumelha, J., Enfield, K.S.S., Almagro, J., Cha, H., Pich, O., Karasaki, T., Moore, D.A., Salgado, R., Sivakumar, M., Young, G., Molina-Arcas, M., de Carné Trécesson, S., Anastasiou, P., Fendler, A., Au, L., Shepherd, S.T.C., Martínez-Ruiz, C., Puttick, C., Black, J.R.M., Watkins, T.B.K., Kim, H., Shim, S., Faulkner, N., Attig, J., Veeriah, S., Magno, N., Ward, S., Frankell, A.M., Al Bakir, M., Lim, E.L., Hill, M.S., Wilson, G.A., Cook, D.E., Birkbak, N.J., Behrens, A., Yousaf, N., Popat, S., Hackshaw, A., Hiley, C.T., Litchfield, K., McGranahan, N., Jamal-Hanjani, M., Larkin, J., Lee, S., Turajlic, S., Swanton, C., Downward, J. and Kassiotis, G. (2023) ‘Antibodies against endogenous retroviruses promote lung cancer immunotherapy’, Nature (London), .
Ou, H. and Schumacher, B. (2018) ‘DNA damage responses and p53 in the aging process’, Blood, 131(5), pp. 488-495.
Quaranta, V., Rainer, C., Nielsen, S.R., Raymant, M.L., Ahmed, M.S., Engle, D.D., Taylor, A., Murray, T., Campbell, F., Palmer, D.H., Tuveson, D.A., Mielgo, A. and Schmid, M.C. (2018) ‘Macrophage-derived granulin drives resistance to immune checkpoint inhibition in metastatic pancreatic cancer’, Cancer Research (Chicago, Ill.), 78(15), pp. 4253-4269.
Sharma, P., Mehta, M., Dhanjal, D.S., Kaur, S., Gupta, G., Singh, H., Thangavelu, L., Rajeshkumar, S., Tambuwala, M., Bakshi, H.A., Chellappan, D.K., Dua, K. and Satija, S. (2019) ‘Emerging trends in the novel drug delivery approaches for the treatment of lung cancer’, Chemico-Biological Interactions, 309, pp. 108720.
Sung, H., Ferlay, J., Siegel, R.L., Laversanne, M., Soerjomataram, I., Jemal, A. and Bray, F. (2021a) ‘Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries’, CA: A Cancer Journal for Clinicians, 71(3), pp. 209-249.
Sung, H., Ferlay, J., Siegel, R.L., Laversanne, M., Soerjomataram, I., Jemal, A. and Bray, F. (2021b) ‘Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries’, CA: A Cancer Journal for Clinicians, 71(3), pp. 209-249.
Tao, L., Zhang, L., Peng, Y., Tao, M., Li, L., Xiu, D., Yuan, C., Ma, Z. and Jiang, B. (2016) ‘Neutrophils assist the metastasis of circulating tumor cells in pancreatic ductal adenocarcinoma: A new hypothesis and a new predictor for distant metastasis’, Medicine (Baltimore), 95(39), pp. e4932.
Yang, Y., Guo, Z., Chen, W., Wang, X., Cao, M., Han, X., Zhang, K., Teng, B., Cao, J., Wu, W., Cao, P., Huang, C. and Qiu, Z. (2021) ‘M2 macrophage-derived exosomes promote angiogenesis and growth of pancreatic ductal adenocarcinoma by targeting E2F2’, Molecular Therapy, 29(3), pp. 1226-1238.
